nf-core/genomicrelatedness
Bioinformatics pipeline for estimating genetic relatedness from low-coverage whole-genome sequencing (sWGS) data
Introduction
nf-core/genomicrelatedness is a bioinformatics pipeline for estimating genomic relatedness from low-coverage whole-genome sequencing (lcWGS) data. It performs read mapping, optional base quality score recalibration, variant calling with GATK and BCFtools, and downstream relatedness estimation. For many non-model organisms, no high-confidence variant set is available. The pipeline provides an automated multi-round bootstrapping workflow to generate one. The resulting standardized outputs include genotype likelihood-based variant calls, filtered VCF files, and relatedness estimates from several independent algorithms, enabling robust inference even from very sparse sequencing data.
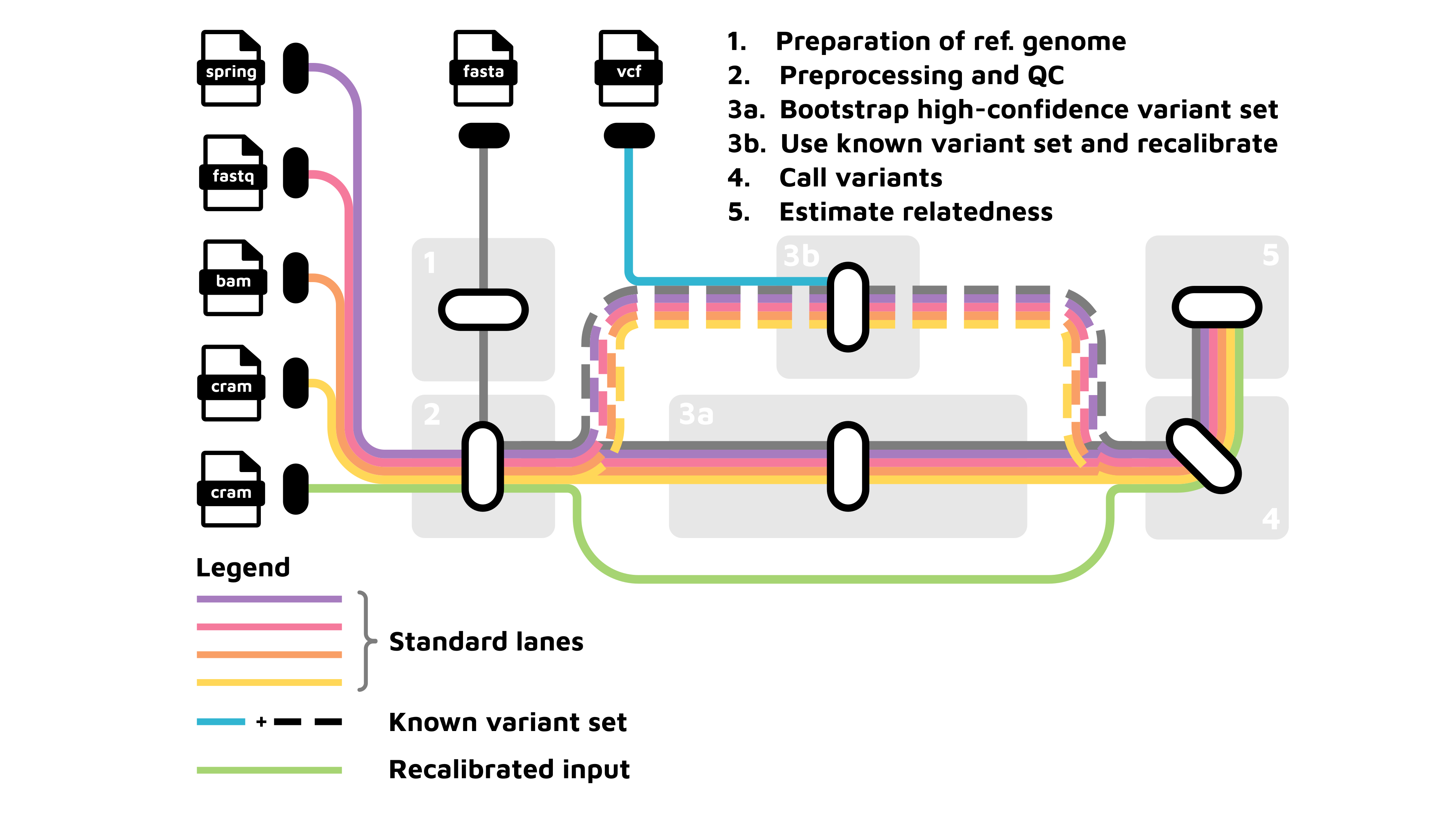
The pipeline consists of the following four main sections that perform the major processing steps:
-
Preprocessing section: Prepares the input files for downstream analyses.
-
Reference genome preparation (
--prepare_genome): This step prepares the input reference genome and generates the relevant files for subsequent processing steps. Key processes include:-
Indexing of the reference genome with
BWA-mem2 -indexto generate alignment index files. -
Indexing of the reference genome with
samtools -faidxto generate FASTA index (.fai) -
Creating sequence dictionary with the
GATK4 -createsequencedictionaryto generate sequence dictionary file (.dict)
-
-
Interval preparation (—prepare_intervals): This step splits the indexed reference genome into intervals for computational more efficient downstrean analyses.
- Build and split intervals of the reference genome with [
gawk -build_intervals] and [split_intervals] to generate interval file (.bed)
- Build and split intervals of the reference genome with [
-
Preprocessing of raw sequencing reads (
--preprocess): This step performs all essential steps to provide aligned sequence reads and quality metrics.-
Input parsing & metadata setup: Reads a CSV samplesheet describing the input FASTQ or SPRING files with raw sequencing reads and their read-group information.
-
Raw read quality control: performs quality control, trimming, filtering and merging of paired reads with
fastp, generating trimmed and filtered paired reads (.merged.fastq.gz) -
Read mapping: Aligns the paired reads to the reference genome with
BWA-mem2 -mem, sorts reads withsamtools -sort, adds read group information withGATK4 -addorreplacereadgroups. -
Merge files stemming from the same sample with
samtools -merge. -
Remove duplicates with
GATK4 -markduplicates. -
Indexing of the mapped reads with
samtools -index, providing sorted and indexed compressed alignment files with proper read group annotations (.cram) and corresponding index files (.crai). -
Quality metrics of the sequencing data are compiled with
MultiQCand include overviews of library complexity analysed withpreseq -c_curveandpreseq -lcextrap, read mapping statics over the various preprocessing steps withsamtools -stats, and genome coverage usingmosdepth.
-
-
-
BQSR section: corrects systematic errors introduced during sequencing by adjusting base quality scores.
-
Variant bootstrapping (
--bootstrap_variant_set): (OPTIONAL) If no known variant set is available, this step automatically generates one via bootsrapping. It iteratively refines and stabilises the set of high-confidence variants for downstream use.-
Variant calling performs joint variant discovery for all samples with
GATK4 -HaplotypeCaller,GATK4 -GenomicsDBImport,GATK4 -GenotypeGVCFs,GATK4 -mergevcfs. -
Hard filtering of variants to create a high confidence reference variant set (.vcf) with
GATK4 -VariantFiltration(Qual >=100, QD < 2.0; MQ < 35.0; FS >60; HaplotypeScore > 13.0; MQRankSum < -12.5; ReadPosRankSum < -8.0) andGATK4 -SelectVariants.
-
-
BQSR (
--base-quality-score-recalibration): uses the produced or a provided variant set to adjust quality scores in alignment files:-
BQSR with
GATK4 -BaseRecalibrator,GATK4 -GatherBQSRReport,GATK4 -ApplyBQSR,samtools -merge, andsamtools -indexto produce recalibrated alignment files (.cram). -
BQSR diagnostics with
GATK4 -AnalyzeCovariatesto evaluate the effectiveness of recalibration. -
Iteration (DEFAULT: 2 rounds) of recalibration.
-
-
-
Genotyping section: calls variants with different algorithms to produce genotype likelihoods for all samples. The default is that both subworkflows run in parallel, but can be adjusted to one or the other.
-
Variant calling with GATK (
--call_variants-gatk) performs joint variant discovery for all samples withGATK4 -HaplotypeCaller,GATK4 -GenomicsDBImport,GATK4 -GenotypeGVCFs,GATK4 -mergevcfs. -
Variant calling with BCFtools (
--call_variants-bcftools) employsbcftools -mpileupandbcftools -callper scaffold, and concatenates results into a full cohort VCF withbcftools -concat. -
Intersection and filtering of variants (
--vcf_intersection_thinning) usesbcftools -isecto retain only variants called by both algorithms. Subsequently, variants are filtered usingvcftools -exclude(for example just retaining autosomal variants using the OPTIONAL parameterinclude_scaffoldsorexclude_scaffolds) andvcftools -thin(DEFAULT —remove-filtered-all –remove-indels –maf 0.025 –recode –recode-INFO-all –max-missing 0.75) to produce the final variant set (.vcf). -
Variant statistics are produced for each subworkflow to judge the quality of the called variants.
-
-
Relatedness estimation section: produces robust estimates of pairwise relatedness suitable for lcWGS data.
- Maximum-likelihood estimation of relatedness from genotype likelihoods with NGSrelate (
--relatedness_ngsrelate) uses genotype likelihoods to infer IBD with maximum-likelihood analysis as implemented inNgsRelatev2.
- Maximum-likelihood estimation of relatedness from genotype likelihoods with NGSrelate (
-
MultiQC reporting: Aggregates quality metrics across all workflow stages into a single interactive report.
For detailed instructions, please refer to the usage documentation.
Usage
If you are new to Nextflow and nf-core, please refer to this page on how to set-up Nextflow. Make sure to test your setup with -profile test before running the workflow on actual data.
First, prepare a samplesheet with your input data that looks as follows:
samplesheet.csv:
sample,fastq_1,fastq_2,RGID,RGLB,RGPL,RGPU,RGSM
sample-1,sample1_S1_L002_R1_001.fastq.gz,sample1_S1_L002_R2_001.fastq.gz,FC1_L002,lib1a,Illumina,FC1_L002_sample-1-a,sample-1-a
sample-1,sample1_S1_L003_R1_001.fastq.gz,sample1_S1_L003_R2_001.fastq.gz,FC1_L003,lib1b,Illumina,FC1_L003_sample-1-b,sample-1-b
sample-2,sample2_S1_L004_R1_001.fastq.gz,sample2_S1_L004_R2_001.fastq.gz,FC1_L004,lib2,Illumina,FC1_L004_sample-2-a,sample-2-a
The sample sheet is provided as a comma-separated value (CSV) file, with one line corresponding to one paired-read set and eight defined columns. The first column “sample” holds the individual name enabling cross-referencing to other datasets for downstream analyses (may refer to individual or sample depending on the unit of interest). Columns “fastq_1” and “fastq_2” hold the filepaths to the paired-end sequencing raw reads for forward and reverse read, respectively. The following five columns give more details on the production of the sequencing data based on SAM/BAM file format specification (The SAM/BAM Format Specification Work…), required for the preprocessing section by the GATK4 (McKenna et al. 2010; Van der Auwera and O’Connor 2020; GATK 2024): “RGID” holds the unique run identifier, e.g. {FLOWCELL}.{LANE}; “RGLB” holds the library identifier; “RGPL” holds the sequencing technology or platform, e.g. ILLUMINA; ”RGPU” holds the platform unit, e.g. {FLOWCELL}.{LANE}.{SAMPLE};”RGSM” holds the individual sample name (hence can equal the “ID” column but might deviate if multiple samples of the same individual are analyzed). Optionally, more columns can be added, for example containing sex and group information.
Alternatively, the samplesheet can be filled with fastq files encoded in SPRING format, or the preprocessing steps can be skipped entirely when BAM or CRAM files are provided.
Now, you can run the pipeline using:
nextflow run nf-core/genomicrelatedness \
-profile <docker/singularity/.../institute> \
--input samplesheet.csv \
--fasta <REFGENOME>
--bootstrapping_rounds 1\
--outdir <OUTDIR>Note: If the parameter
--bootstrapping_roundsis provided, it must be an integer between 1 and 3.
Please provide pipeline parameters via the Nextflow -params-file option. Custom config files including those provided by the -c Nextflow option can be used to provide any configuration except for parameters; see docs.
{
"email": "your@email.adress",
"email_on_fail": "your@email.adress",
"input": "./samplesheet.csv",
"fasta": "REFGENOME.fna",
"bootstrapping_rounds": 1,
"target_number_of_interval_files": 150,
"include_scaffolds": "./scaffolds_include.txt",
"skip_relatedness_estimation": true
}For more details and further functionality, please refer to the usage documentation and the parameter documentation.
Pipeline output
The output of the preprocess subworkflow consists of comprehensive quality control reports via MultiQC, including metrics on library complexity, coverage, and duplication rates, and the CRAM files ready for the subsequent analysis steps. The BQSR workflow produces diagnostic plots to evaluate the effectiveness of recalibration and the recalibrated CRAM files for further analyses. The output of the genotyping section includes the intermediary and final variant set. The output of the relatedness estimation section is the pairwise relatedness matrices. To see the results of an example test run with a full size dataset refer to the results tab on the nf-core website pipeline page. For more details about the output files and reports, please refer to the output documentation.
Credits
nf-core/genomicrelatedness was originally written by Thomas Isensee in collaboration with Gisela H. Kopp and Till Dorendorf. This work was carried out as part of the bwRSE4HPC initiative, funded by the Baden-Württemberg Ministry of Science, Research and Arts, coordinated by the Scientific Software Center (SSC) at Heidelberg University and the Scientific Computing Center (SCC) at KIT.
We thank the following people for their extensive assistance in the early stages of the development of this pipeline:
Contributions and Support
If you would like to contribute to this pipeline, please see the contributing guidelines.
For further information or help, don’t hesitate to get in touch on the Slack #genomicrelatedness channel (you can join with this invite).
Citations
An extensive list of references for the tools used by the pipeline can be found in the CITATIONS.md file.
You can cite the nf-core publication as follows:
The nf-core framework for community-curated bioinformatics pipelines.
Philip Ewels, Alexander Peltzer, Sven Fillinger, Harshil Patel, Johannes Alneberg, Andreas Wilm, Maxime Ulysse Garcia, Paolo Di Tommaso & Sven Nahnsen.
Nat Biotechnol. 2020 Feb 13. doi: 10.1038/s41587-020-0439-x.